2025-12-29
重组腺相关病毒(rAAV)载体因其显著优势,已成为体内基因治疗领域的首选病毒载体。相较于其他病毒载体,rAAV具有物理化学性质稳定、致病性低、整合风险小以及转基因长期表达等优势。近年来,获批上市及开展临床试验的rAAV产品数量快速增长,为难治性和罕见病治疗带来了希望。然而,rAAV产品具有高度创新性和复杂性,其生产工艺多样且持续改进。与此同时,质量控制方法和技术也在快速进步。随着生物技术的快速发展,监管机构与申请rAAV产品用于人体试验或商业化的实体之间加强沟通的需求日益迫切。
近日国家药品监督管理局药品审评中心(CDE)发布了一篇题为Regulatory Perspective for Biologics License Application of Recombinant Adeno-Associated Virus Products in China的文章,重点探讨在我国申报rAAV产品监管批准时涉及的关键化学、生产及控制(CMC)方面的问题,包括不同生产工艺中外源病毒的控制以及rAAV产品生物制品许可申请过程中的质量控制,为rAAV的生产与质量控制提供参考依据和建议,从而加速基因治疗产品的高质量开发。
劲帆生物医药科技(武汉)有限公司简称“劲帆医药”,创立于2022年,提供一站式病毒载体、蛋白和疫苗CRO/CDMO服务,拥有全球领先的规模化AAV制备专利技术平台及病毒载体/cGMP级蛋白/疫苗生产车间,致力于推进基因治疗药物、蛋白药物、治疗性疫苗更有效,更安全,更经济,更可及,赋能客户,造福患者。劲帆医药cGMP生产车间,包括4个P2级别种子建库区、5条蛋白/病毒生产线和1条灌装生产线,蛋白/病毒生产线生产规模分别为200L和500L,年最大产能可达50批次,目前已完成多项项目交付。
我国针对先进疗法医疗产品的CMC指南
中国先进治疗药物的研发正快速发展,自2018年以来,上市申请和临床试验申请数量持续增长。随着基因递送载体和基因编辑工具等生物技术的快速发展,先进治疗药物为许多罕见遗传病和常见难治性疾病带来了新希望,并在临床试验中不断取得进展。过去几年间, 国家药品监督管理局(NMPA)陆续发布了系列技术指南,有效规范和指导先进治疗药物的研发、生产和应用(表1)。这些药学生产质量管理规范(CMC)指南建立了一套有利于中国先进治疗药物发展的监管体系。
表1 中国先进治疗用药品的化学、生产与控制指南
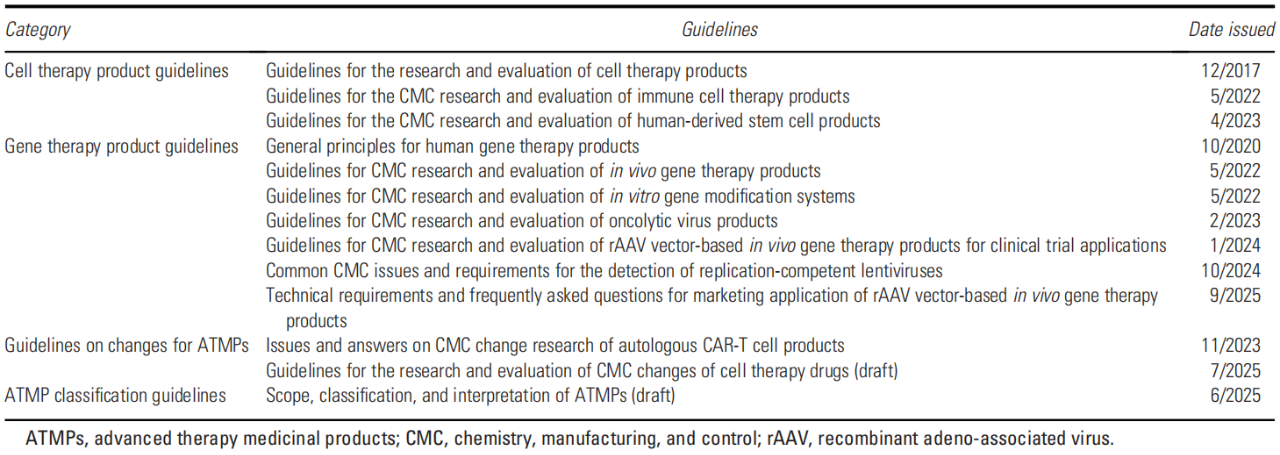
(表格来源:Jing Cui et al., Hum Gene Ther., 2025)
重组AAV产品的CMC挑战和监管考虑
根据目前中国CMC研究和rAAV产品应用的现状,主要挑战总结如下:(1)生产工艺的多样性,包括细胞系的选择和辅助病毒的引入,使得不同机构之间以及针对所生产的各种载体难以标准化制造流程。(2)不同制造工艺引入的病毒成分程度各异,这使得难以建立行业标准来控制外源病毒,导致产品和污染物评估的多样性。(3)所生产的rAAV载体以及确定产品强度和纯度的方法都在快速迭代改进,因此建立和验证标准化质量控制方法面临重大挑战。(4)rAAV产品含有多种复杂的工艺和产品相关杂质,如空衣壳或部分填充衣壳、残留质粒DNA和复制型AAV(rcAAV)。然而,此类杂质对rAAV载体安全性的影响尚不明确,且国际监管指南中尚未对产品中杂质限度制定统一标准。(5)效力测定是产品特性分析、可比性研究及稳定性方案中不可或缺的环节,这些测试旨在确保临床研究各阶段的产品生产质量始终如一。然而,由于基因治疗产品的作用机制复杂,单一生物学或分析检测方法可能无法准确评估其效力。因此,若采用通用标准化方案而非针对具体基因治疗产品制定的方案,可能会遗漏生产批次间存在的关键差异。
不同生产环境中外源病毒的监管考量
目前,有三种常用的rAAV制造平台已经经历了显著的发展和优化:(1)HEK293细胞中的三质粒瞬时共转染系统。(2)Sf9细胞中的杆状病毒-昆虫细胞系统。(3)辅助病毒生产系统。表2对比了三种平台生产的rAAV产品在可扩展性、监管挑战、病毒安全性风险及商业适用性方面的表现。
表2 三种主要重组腺相关病毒生产平台的比较
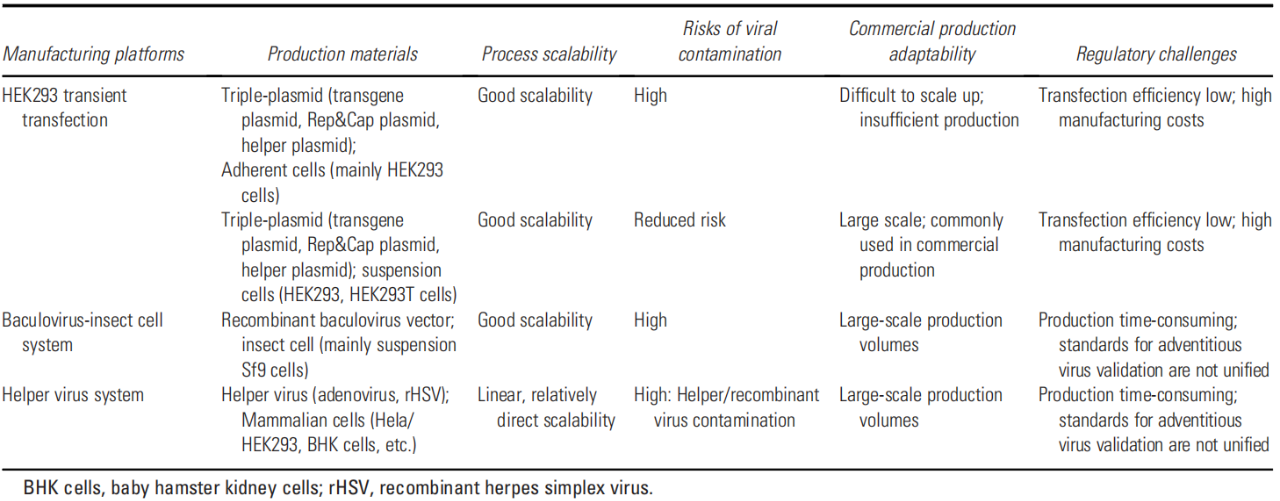
(表格来源:Jing Cui et al., Hum Gene Ther., 2025)
病毒污染是所有AAV产品生产过程中固有的潜在风险。国际协调理事会(ICH)Q5A(R2)监管指南提出了控制生物技术产品病毒污染的三种主要互补的方法:(1)筛选并检测细胞系及其他原材料(包括培养基成分),以确保其不含不良传染性病毒。(2)评估各生产工艺引入明确外源性和内源性病毒的风险。(3)采取相应的生产步骤检测产品,以确保生产过程中不混入污染性传染病毒。但目前国际监管机构尚未就rAAV产品病毒清除验证的要求达成共识,且全球监管机构也未针对不同类型生产方式制备的rAAV产品制定外源病毒整体风险控制策略的具体要求。CDE针对上述三种不同rAAV生产工艺提出外源病毒控制策略,并详细阐述了技术要求。具体要求详见图1及后续内容。
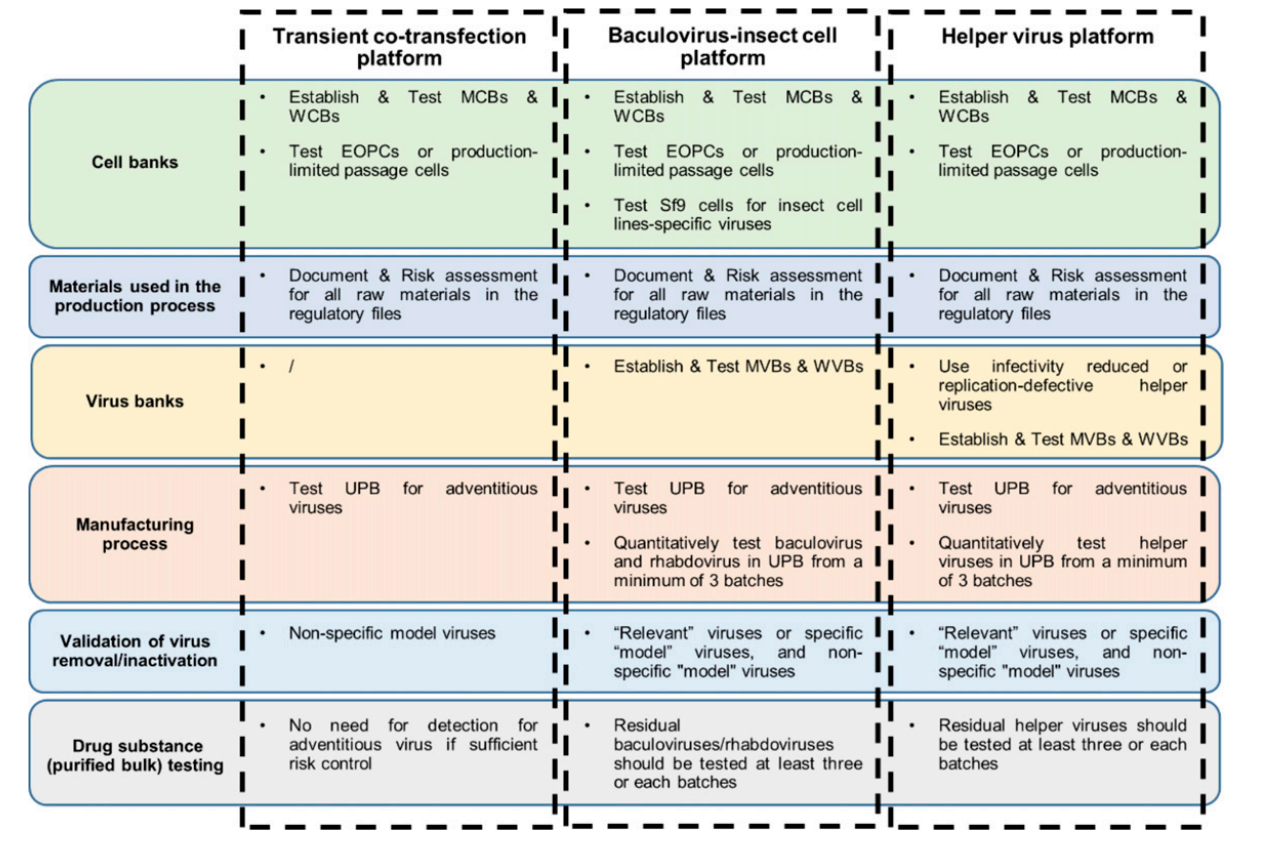
图1 主要生产平台的外源病毒控制工作流程
(图片来源:Jing Cui et al., Hum Gene Ther., 2025)
一、瞬时共转染平台
目前最广泛采用的生产方法是HEK293细胞的三质粒共转染技术。其主要优势包括生产过程早期开发周期相对较短、质粒生产、制备和质量控制方法相对简单,且无需引入辅助病毒,降低了外源病毒的风险。尽管该方法不会主动引入潜在外源病毒,但这些病毒可能源自生产细胞系、生产过程中使用的动物/人源原材料,或制造环节的污染。
1. 细胞库中外源病毒的控制
HEK293细胞广泛用于生物制品生产,因此建立无病毒污染的细胞库已成为普遍实施的控制流程。为最大限度降低HEK293细胞污染风险,建议使用来源明确的细胞,并以标准化方式建立细胞库,例如主细胞库(MCBs)和工作细胞库(WCBs)。此外,生产终末细胞或生产受限传代细胞应进行污染检测。
2. 材料中外源病毒的控制
培养基和原材料的具体评估应记录在监管文件中,涉及成分、纯度、无菌性、生物活性及外源因子缺失情况。通常,rAAV产品生产采用化学定义的无血清培养基,动物源性组分包括胰蛋白酶和血清。对于所有动物源性组分,应记录其来源生物体、供应商/厂商、原产国、病原体检测结果及使用阶段等信息。收集这些信息旨在降低牛海绵状脑病、传染性海绵状脑病等疾病传播及病原体传播的潜在风险;因此应采用基于风险的方法评估rAAV产品生产中所用原材料引入外源因子及其他污染物的可能性。
3. 生产工艺中外源病毒的控制
在rAAV产品的生产过程中,材料添加或转移环节可能引入来自人类或环境的病毒污染物。因此,必须维持严格控制和监测的环境,同时确保更衣操作和无菌技术的正确使用。与此同时,还需严格管控生产工艺流程,选择合适的生产步骤进行外源病毒检测,并制定验收标准。未处理原液(UPB)的代表性样本(即在开始加工步骤前从生产反应器中取出的样本)是检测外源病毒污染最适宜的样本水平之一。因此,应对UPB进行适当的病毒检测。
4. 病毒去除/灭活验证
在适当情况下,可将灭活和/或清除步骤整合至生产流程,以进一步降低意外病毒污染的风险。从控制产品病毒安全性的角度出发,所有产品均需进行病毒清除验证;这是因为生产设施可能无法完全避免外源病毒的意外引入,或同一生产场所采用其他工艺时存在的病毒污染风险。若生产工艺设计中包含病毒清除和/或灭活步骤,将能为产品的病毒安全性提供更有力的保障。此外,若在细胞库或UPB中未检出除预期产品外的病毒、类病毒颗粒或类逆转录病毒颗粒,则可采用非特异性模型病毒进行病毒清除验证。若未检出类逆转录病毒颗粒且产品增强型逆转录酶检测结果为阴性,则无需对每剂次的逆转录病毒颗粒进行估算。
5. 产品中外源病毒的检测
如前所述,应在整个生产过程中采取一系列措施以控制外源病毒污染的风险。总体而言,外源病毒污染的风险评估被认为可控,因此无需对原料药(纯化原液)进行外源病毒检测。
二、 杆状病毒-昆虫细胞平台
相较于瞬时共转染法,杆状病毒-昆虫细胞系统在生产规模上具有显著优势。该系统不仅常采用无血清培养基,还成功应用于酶制剂、糖蛋白、重组病毒及疫苗等多种产品的生产,这些产品均展现出良好的安全性特征,使得该系统与其他rAAV生产方法的整合应用更为便捷。不过,由于生产过程中需使用重组杆状病毒和昆虫细胞,该系统需要制定完善的外源病毒防控策略。
1. 细胞库与材料中外源病毒的控制
杆状病毒-昆虫细胞生产系统中细胞库及原料的病毒污染风险控制要求与瞬时共转染平台类似。在检测Sf9细胞污染时,需重点检测已报道的特定病毒(如结节病毒)以及可能在昆虫细胞系中持续存在的对人类有感染性的病毒。此外,部分Sf9细胞可能携带谷蠹杆状病毒(Spodoptera frugiperda rhabdovirus)的基因,因此应考虑进行特异性杆状病毒检测。
2. 病毒库中外源病毒的控制
建立病毒库系统对于维持rAAV生产的一致性至关重要。应建立并测试双层病毒库系统(主病毒库和工作病毒库),包括无菌性、支原体及外源病毒等指标。
3. 生产工艺中外源病毒的控制
与瞬时共转染平台相比,杆状病毒-昆虫细胞系统中外源病毒的体外检测更具挑战性,因为UPB中的杆状病毒可能干扰外源病毒的检测。因此建议在检测外源病毒前使用抗体中和UPB中的杆状病毒。若无法获得合适的中和抗体或中和效果不理想,则应使用生产控制细胞进行外源病毒检测。此外,ICH Q5A(R2)指南指出,可考虑采用核酸扩增和二代测序等分子生物学方法检测特定病毒。为获得上市许可,必须对至少三个商业化生产批次的UPB中杆状病毒和杆状病毒科病毒进行定量检测,以评估整个生产工艺对外源病毒的清除效果。
4. 病毒去除/灭活验证
Sf9细胞可能含有杆状病毒,且重组杆状病毒被引入生产系统。因此,根据ICH Q5A(R2)指南建议,必须在生产工艺步骤中纳入病毒去除/灭活步骤,并对病毒去除/灭活进行验证研究。
在病毒清除验证研究中,应选用“相关”病毒(即已确认或可能污染生产流程的病毒)。若无法获取“相关”病毒,或其不适用于病毒清除研究的工艺评估时,应采用特定“模型”病毒作为替代,例如杆状病毒和水泡性口炎病毒(代表Sf9细胞系中的内源性杆状病毒)。此外,应考虑使用对物理或化学处理具有显著抗性的非特异性“模型”病毒作为验证对照,以确保病毒去除/灭活步骤的有效性。模型病毒的类型与数量应根据包装/生产细胞与生产工艺决定。通常建议至少检测三种具有不同特征的病毒,以评估生产工艺的病毒清除能力。
5. 产品中外源病毒的检测
根据检测结果和风险评估,应通过使用“相关”许可细胞系进行感染性检测,对至少三个原料药(纯化原液)批次进行检测,以确认残留杆状病毒和杆状病毒的缺失。也可采用分子检测方法进行检测。除非有充分的过量清除证据支持,否则应对每个纯化原液制剂进行残留杆状病毒和杆状病毒的检测。
三、 辅助病毒平台
尽管腺病毒(AdV)和rHSV等辅助病毒的应用为rAAV复制提供了必要条件,但也带来了外源病毒(包括辅助病毒本身)的潜在风险。因此,有必要建立多阶段风险控制策略。
1. 细胞库与材料中外源病毒的控制
细胞库(MCBs)、外源细胞库(WCBs)及材料中病毒污染的控制要求与瞬时转染平台相同。
2. 病毒库中外源病毒的控制
基因工程技术已被开发用于减弱辅助病毒的感染性或使其复制缺陷,这显著降低了其致病性和自主复制能力,从而降低rAAV产品中的污染风险。为确保安全性,病毒基因组(VG)中通常会删除编码感染细胞蛋白0、4、22、27和47的立早基因。此外,建议建立辅助病毒库系统,并对无菌性、支原体及外源病毒进行特性分析。
3. 生产工艺中外源病毒的控制
必须考虑对UPB进行外源病毒检测,并在检测前使用适当抗体中和辅助病毒。除在生产过程中对产品进行检测外,还应在生产控制细胞或分子生物学方法(如核酸扩增)上进行外源病毒检测,并根据ICH Q5A(R2)指南对特定病毒检测的要求采用新一代测序技术。
4. 病毒去除/灭活验证
ICH Q5A(R2)指南中提供了关于实施去除“相关”病毒工艺的详细要求和建议。简而言之,该工艺应能以高安全裕度证明可去除“相关”病毒,并应包含一个特征明确的灭活步骤。若需通过不同步骤实现病毒清除效果叠加,分离原理必须相互独立。鉴于AAV与Ad5已明确的差异性热稳定性,热灭活可轻松实现Ad5>5-6个对数级的病毒量减少。“纳滤”是目前重组蛋白生产工艺中常规采用的额外病毒清除措施。由于rAAV载体体积较小,可从专为逆转录病毒清除设计的过滤器中定量回收,因此此类过滤器应纳入现行rAAV良好生产流程。
5. 产品中外源病毒的检测
产品中外源病毒的检测要求与杆状病毒-昆虫细胞生产工艺相同,唯一区别在于需检测的病毒类型。对于辅助病毒生产工艺,如rHSV和AdV等辅助病毒可能构成潜在污染风险。
rAAV质量控制的监管考量
一、 强度或滴度
由于载体基因组是rAAV产品治疗效果的关键成分,VG滴度是最常用的确定剂量的关键质量属性(CQA)。患者接受的VG数量对安全性和疗效有重大影响:剂量过低会导致治疗效果有限,而剂量过高则可能因转基因过表达引发不良反应。因此,如何准确测定VG滴度对优化产品的安全性和有效性至关重要。目前业界通常采用定量聚合酶链反应(qPCR)或液滴数字聚合酶链反应(ddPCR)进行检测。近年来,ddPCR因其相较于传统qPCR的多项优势(包括降低样本基质干扰、无需标准曲线即可实现绝对定量,以及更高的检测精度和准确性)已成为行业标准。
随着临床试验的推进,检测方法可能因技术快速发展而发生变更。但建议在临床试验早期阶段就实施方法变更,以避免方法差异成为干扰因素。在临床试验中实施任何VG检测方法变更前,应根据具体变更情况开展全面风险评估,确保新方法在产品质量控制方面与原有方法具有同等或更优的能力,并保证临床剂量的准确性。
二、 病毒蛋白纯度
衣壳蛋白比例的差异会通过影响转导效率,进而改变rAAV的体内效力。检测rAAV衣壳蛋白电荷异质性的方法主要包括十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)和毛细管电泳-十二烷基硫酸钠法(CE-SDS)。在分辨率、精确度和稳健性方面,CE-SDS方法优于SDS-PAGE。随着技术的持续进步,新的改进检测方法可能会在生产过程中开发并应用。因此,建议在产品纯度检测中优先采用精度和准确性更高的检测方法。
在使用CE-SDS法评估AAV843衣壳蛋白纯度时,除了预期的VP1、VP2和VP3亚基在CE-SDS图谱中的峰外,还检测到一个分子量略小于VP3的未知信号峰,推测可能是翻译过程中产生的VP3变体。然而,这种变体如何影响衣壳组装所需蛋白质的化学计量比尚不清楚。建议制定控制策略以量化不同阶段VP1、VP2、VP3及其变体,决定哪些因素可能导致其产生或抑制,并明确其与产品特性(如安全性、有效性及质量控制)的关联。必要时,除需控制rAAV产品中VP1+VP2+VP3的总量外,还应根据VP3变体对病毒结构或功能影响的风险评估,考虑对VP3分子变体含量进行单独控制。
三、 尺寸变化
rAAV产品中单体和聚集体的含量是一个CQA,反映了生产过程的稳健性和产品的质量特性。目前,有几种方法用于检测rAAV产品中的单体和聚集体,例如尺寸排阻色谱(SEC)、分析超速离心(AUC)和动态光散射。SEC根据溶质的流体力学体积分离亚可见尺寸(d < 100 nm)的溶质。它还用于高效液相色谱(HPLC)系统,配备紫外、荧光光谱仪或差示折射率检测器用于定量洗脱物质,因其操作简便、通量高且速度快,已成为评估聚集体的行业标准方法。
虽然基于SEC的检测方法因其操作简便、通量高而长期作为行业标准,但由于存在过滤问题、非特异性相互作用及解聚现象,它们并不适用于大颗粒rAAV聚集体的定量分析。AUC法虽能评估聚集程度,但其通量低且对小聚集体浓度的检测精度不足,成为工艺开发中的制约因素。动态光散射技术作为在线检测手段在大聚集体定量方面展现出潜力,但其精确度较低,且当基质光学特性存在不确定性时可能产生偏差。尽管每种方法各有优缺点,但在重组腺相关病毒产品的研发阶段,仍建议采用多种技术手段对产品单体及聚集体进行全面表征研究。进入临床试验阶段后,需持续监测各批次产品中单体与聚集体的数据变化,以及稳定性期间的动态变化,从而制定出产品中单体与聚集体的标准限值。
四、 杂质
rAAV产品含有多种类型的杂质,包括工艺相关杂质和产品相关杂质。工艺相关杂质源自材料和组件的制造过程,但与产品结构无关。产品相关杂质是指在生产或储存过程中由产品组件产生的非预期、无功能性的产物。本文将讨论残留宿主DNA和空衣壳病毒的质量控制考量。
1. 残留宿主DNA
含有病毒癌基因的细胞常被用于制造AAV载体。例如,含有AdV E1基因的HEK293细胞,以及含有人乳头瘤病毒E6和E7基因的HeLa细胞。NMPA和FDA指南要求基因治疗产品必须控制残留宿主细胞DNA及DNA片段大小,并建议将残留DNA控制在每剂10 ng以下,残留DNA片段大小控制在200 bp以下。此外,对于具有潜在安全风险的产品中已知的转化因子(如E1、E6和E7基因),还应进行残留量检测和控制。这些致癌序列可通过qPCR或ddPCR进行定量检测。与qPCR相比,ddPCR的准确度较低且动态范围较窄,但ddPCR的成本更高。当残留宿主细胞DNA作为核酸酶敏感的工艺杂质存在时,可通过成熟的工艺优化策略满足这些要求。例如,残留宿主DNA可被核酸酶消化成小片段,并通过后续工艺如亲和层析、阴离子交换层析、超滤-渗滤或切向流过滤进行去除。然而,封装在AAV衣壳内的耐核酸酶宿主细胞DNA残留物检测困难且难以清除。据报道,源自载体模板和辅助序列的包装残留DNA杂质在纯化载体颗粒中占总DNA的1%至8%。
2. 空衣壳病毒
rAAV产品通常需要检测三种类型的衣壳:完整型、中间型和空衣壳型。虽然中间型和空衣壳对rAAV产品的安全性和有效性影响尚未完全明确,但它们通常被视为杂质。此外,部分AAV颗粒仅部分包装了目标基因盒,仅包含内含子-外显子重复序列(ITRs)及相邻DNA序列。这些不完整或缺陷的AAV颗粒会增加给药需求,不过这种影响在直接组织给药时可能更为明显,而在全身给药时则相对轻微。值得注意的是,部分衣壳还可能含有非预期的DNA序列,例如截短形式的治疗基因,宿主细胞DNA和/或残留质粒DNA,因此可能由于潜在的遗传毒性或免疫原性效应而构成安全性问题。
目前,检测重组腺相关病毒产品空衣壳率的常用方法包括AUC、离子交换(IE)- HPLC和尺寸排阻色谱-多角度光散射(MALS)。IE- HPLC和SEC- MALS方法平台成熟,分析速度快且通量高,但可能无法完全区分部分包装和完全包装的衣壳。与IE- HPLC和SEC- MALS相比,AUC对完全包装、部分包装和空衣壳病毒颗粒具有更好的分离效率,测量的空衣壳率与理论空衣壳率之间的差异较小,因此在工业应用中广泛使用。随着技术的不断进步和发展,新的、更好的检测方法可能会逐渐应用。因此,鼓励优先采用高精度和高准确性的检测方法来检测和控制空衣壳率。
NMPA 、FDA和EMA指南均强调控制空衣壳病毒的必要性,但均未能明确提供针对特定检测方法和标准限度的具体要求。制定空衣壳率标准限度的关键考量因素包括产品特性、生产工艺、历史批次检测数据、给药方式与剂量,以及与临床安全性和有效性的相关性。同样地,作为VG测量指标,若产品临床试验期间空衣壳率的检测方法发生变更,需评估该检测方法变更对安全性和有效性结果的影响。总体而言,该领域亟需合理制定空衣壳率的标准限度,既适用于通用情况,也适用于各具体产品。
3. 效力评估
rAAV产品的效力检测可包括测定产品浓度(亦称强度或滴度)及功能活性的检测方法。如前所述,对于大多数rAAV产品而言,VG滴度是决定剂量的关键检测指标。然而,必须开发新的方法来测量载体的功能活性,并检测载体基因组向靶细胞的递送情况(感染性)、转基因产物的后续表达,以及转基因产物的实际功能活性。
在上市应用阶段,效力标准限度应基于产品开发、生产及临床结果中积累的数据设定,并根据临床试验数据、方法学变异性、批次间差异及稳定性期间的变化进行修订。若在产品研发过程中对效力评估方法进行修改,应结合内容、范围及变更风险提供充分的风险评估与研究数据,以判定其是否对产品效力测定产生不利影响。
4. 安全性
此类检测应包括无菌性、支原体、内毒素、通用安全性及外源病毒的检测项目。必须对药品进行无菌性检测,且该检测需通过评估载体产品的抑菌和抗真菌活性来验证其有效性。内毒素检测应采用经验证的检测方法进行,其验收标准必须适用于计划的给药途径。
由于生产过程中可能发生的自发重组事件,病毒载体可能获得复制能力,从而增加免疫毒性风险。其机制主要被认为是在生产过程中,载体基因组中的元件与包装质粒上的AAV rep基因和cap基因序列发生非同源重组。临床rAAV产品需进行质量控制检测,推荐采用基于细胞的检测方案:在Ad5存在条件下进行多轮转导以扩增低丰度病毒信号,随后通过qPCR检测rep基因。使用该方法检测rcAAV时,需重点考虑阳性对照病毒的选择、共培养细胞的筛选以及标准限值的建立。具体而言,为验证检测方法的灵敏度和准确性,建议阳性对照病毒的血清型应与产品血清型保持一致。此外,鉴于不同血清型病毒在不同细胞中的感染特性或能力存在差异,建议筛选对每种产品最易感的细胞系。
目前针对rcAAV的标准限值尚未形成监管共识。已进入临床试验阶段的rAAV产品scAAV2/8-LP1-hFIXco,采用细胞培养与定量PCR联用技术进行检测,可将rcAAV控制在每2.25×106 VG≤1个颗粒。目前使用的载体生产系统已优化至能将rcAAV控制在每1×108 VG≤1个颗粒。这些报告数值部分取决于检测rcAAV的分析方法灵敏度极限——这类方法通常依赖于在腺病毒(AdV)存在下转染rAAV产品后,对允许性细胞系进行多轮传代扩增AAV rep基因和/或cap DNA序列。然而,监管机构并未规定判断产品是否含有rcAAV的具体数值阈值。相反,建立rcAAV检测限与耐受性的标准限值合理性需通过综合评估基于PCR的检测方法、累积批次数据以及临床安全性和有效性等因素来确定。
结论
由于rAAV生产中的病毒清除步骤可能无法达到重组蛋白生产的同等稳健性,因此必须通过针对产品定制的风险评估来确保其病毒安全性——这三种成熟的rAAV生产方法(三重共转染、昆虫杆状病毒和辅助病毒)各自存在特定风险。风险评估需考量特定CQA,同时需要识别更多适用于rAAV产品的相关CQA并以更高灵敏度进行检测。随着技术进步,基因治疗产品的制造工艺将持续实现迭代优化与创新突破,推动遗传医学领域快速演进。因此,对CAQ检测方法和标准检出限和耐受性的考虑也将不断发展,以制定更好的非定病毒控制策略,使rAAV产品更安全有效。
劲帆医药严格遵循国家生物制品生产质控标准,以专业的服务和精益求精的态度,为每个客户提供高质量、专业化的CDMO服务。我们将持续探索并优化AAV相关的CDMO工艺,以更优、更快、更具成本效益的技术,满足AAV药物研发与生产的CDMO需求。
参考文献:
https://www.liebertpub.com/doi/10.1177/10430342251393707?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub++0pubmed

地址:中国武汉东湖高新区光谷七路128号 市场:17720522078 人事行政:027-62439686 邮箱:marketing@genevoyager.com
BD 商务总台:13886000399(BD 经理)
本公司所有产品仅供实验科研使用,不用于人体疾病治疗及临床诊断。
地址:中国武汉东湖高新区光谷七路128号 市场:17720522078 人事行政:027-62439686 邮箱:marketing@genevoyager.com
BD 商务总台:13886000399(BD 经理)
本公司所有产品仅供实验科研使用,不用于人体疾病治疗及临床诊断。
